Targeted Proteomics Data
AMP PD features two separate Targeted Proteomics datasets and one Targeted Proteomics release product. AMP PD Release 4.0 contains a bridged data release product generated from Release 3.0 datasets, containing samples from the NINDS Parkinson’s Disease Biomarkers Program (PDBP) and Michael J Fox Foundation’s Parkinson’s Progression Markers Initiative (PPMI).
Release 3.0 contains two datasets generated using Olink 1536 from two separate data providers, labeled ‘D01’ and ‘D02’. Within the dataset ‘D01’, there were 746 CSF and Plasma samples from 213 participants. Dataset ‘D02’ includes 666 CSF samples & 898 plasma samples from 225 participants matched across both tissue types. This results in a total of 3050 samples from 413 participants.
These Targeted Proteomics datasets and release product contain eight unfiltered NPX files from four separate Panels for both Plasma and CSF samples. The four targeted proteomics panels are Cardiometabolic, Inflammation, Neurology, and Oncology.
Release 4.0 contains a bridged data release product that was generated using Olink provided bridging scripts in R. This is available under the Release 4.0 tag within the proteomics folder. In Release 3.0, all datasets are listed under the main datasets tab within the proteomics folder. All data products are split into four separate datasets. The data is split by tissue source (CSF, Plasma), data set number (D01, D02) and release (D03). From there, the main folders for each data set contain data from all four panels in both long and matrix format. An additional format is provided in the olink-explore-format folder. This format is provided for those who wish to use Olink specific tools. Each dataset also contains a sample metadata sheet for user reference.
Data has been reformatted from the original format to contain AMP PD specific participant and sample IDs. Additional QC columns have been added to the data in order to provide additional information for flagged or passed samples. The released data has accompanying Terra notebooks, ranging from “Getting Started” notebooks which help users get familiar with the data and eventually assign case and control to samples, to QC notebooks to show users QC criteria used in flagging samples and analysis notebooks to perform simple data visualizations.
As AMP PD accumulates datasets of the same source tissue and compatible methods, updates to the normalized aggregate release product will be made. Datasets in the datasets directory should not be combined in researchers' analyses without first assessing compatibility and normalizing their values, therefore, users wishing to use NPX values from both ‘D01’ and ‘D02’ datasets should use the Release 4.0 Proteomics Release product.
What's on this page:
| Baseline | 3M | 6M | 9M | 12M | 18M | 24M | 30M | 36M | 42M | 48M | 54M | 60M | 72M | 84M | 96M | Total | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDBP | Plasma | 111 | 0 | 2 | 0 | 91 | 28 | 114 | 0 | 110 | 0 | 48 | 0 | 17 | 0 | 0 | 0 | 521 |
| CSF | 111 | 0 | 2 | 0 | 91 | 28 | 114 | 0 | 110 | 0 | 48 | 0 | 17 | 0 | 0 | 0 | 521 | |
| PPMI | Plasma | 263 | 26 | 125 | 9 | 128 | 24 | 166 | 26 | 94 | 18 | 148 | 15 | 55 | 7 | 5 | 1 | 1120 |
| CSF | 244 | 5 | 111 | 0 | 109 | 0 | 152 | 1 | 84 | 0 | 133 | 1 | 43 | 0 | 5 | 1 | 888 |
Sample Selection Criteria
The criteria used to select these samples were as follows:
- Samples for three timepoint or more available
- Participant samples selected were from participants who had previously generated corresponding Whole Genome Sequencing or Transcriptomic data on the AMP PD Knowledge Platform
- All CSF samples selected had hemoglobin < 100 ng/mL to assure limited blood contamination
- All Plasma and CSF samples were collected under similar protocols
AMP PD Quality control of the preview release data was performed by Victoria Dardov from Technome as part of a contract with the Foundation for the National Institutes of Health (FNIH).
Information here was prepared by the Olink Proteomics in consultation with the AMP PD Proteomics Working Group.
Method
{"preview_thumbnail":"/sites/default/files/styles/video_embed_wysiwyg_preview/public/video_thumbnails/UFvPNPyueNc.jpg?itok=NqCC4IQa","video_url":"https://youtu.be/UFvPNPyueNc","settings":{"responsive":1,"width":"854","height":"480","autoplay":1},"settings_summary":["Embedded Video (Responsive, autoplaying)."]}
Proximity Extension Assay for Targeted Proteomics

Normalized Protein Expression (NPX) quantifies the relative amount of a specific protein. This is determined by performing an immunoassay for a targeted protein. Antibodies that bind to a protein of interest contain unique sequences that are extended, amplified and subsequently detected and quantified by NGS. The amount of this sequence is normalized to standard plate controls to give relative quantities of targeted proteins.
Assay Controls Summary
Extensive quality control is performed for each assay in order to control and assess technical performance of the assay at each step. This ensures generation of reliable data.
AMP PD Controls
AMP-PD quality control further examines the data and includes sample, run and control sample QC.
Generating NPX values
The Explore system´s raw data output are NGS counts, where each combination of an assay and sample is given an integer value based on the number of DNA copies detected. These raw data counts are converted into NPX values for use in downstream statistical analysis.
Assay Controls
Converting Counts to NPX
The Explore system´s raw data output are NGS counts, where each combination of an assay and sample is given an integer value based on the number of DNA copies detected. These raw data counts are converted into NPX values for use in downstream statistical analysis.
NPX generation and Normalization
The NPX values are calculated in two main steps and then intensity normalized by performing between-plate-normalization. First, the assay counts of a sample are divided by those of the extension control for that sample and block, which then undergoes log2 transformation to normalize the data:
Steps in the NPX generation described in equation form, where “i” refers to a specific assay, j refers to a sample, and ExtNPX defines an extension normalized NPX value.
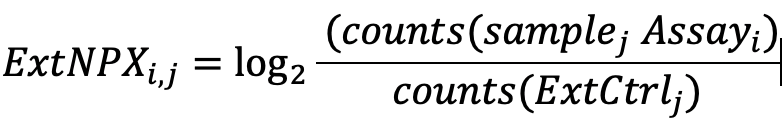
- Relate counts to a known standard (Extension control)
- For all assays and all samples, including negative controls, Plate Controls, and Sample controls.
- Log2 transformation gives more normally distributed data
The result is a scale that has increasing sample values against increasing protein concentration for each assay. The median of the Plate Control is then subtracted from the normalized data:
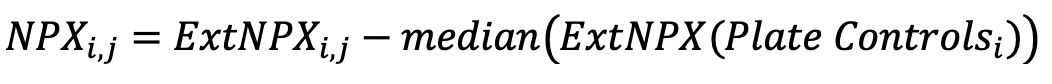
- Perform plate standardization
- For all assays and per plate of samples
Intensity normalization of data is the default for randomized studies on multiple sample plates. In this setting, the median for a random selection of samples is more stable across plates than the three PC’s on each plate. Intensity normalization sets the median level of all assays to the same value for all plates:
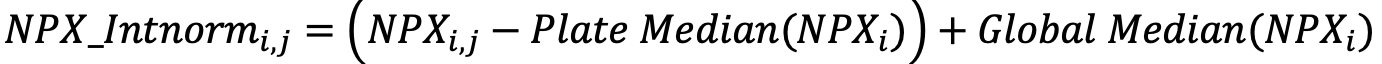
- Between plate normalization (Optional but the default for multi-plate projects)
- For each assay, for all plates in the project.
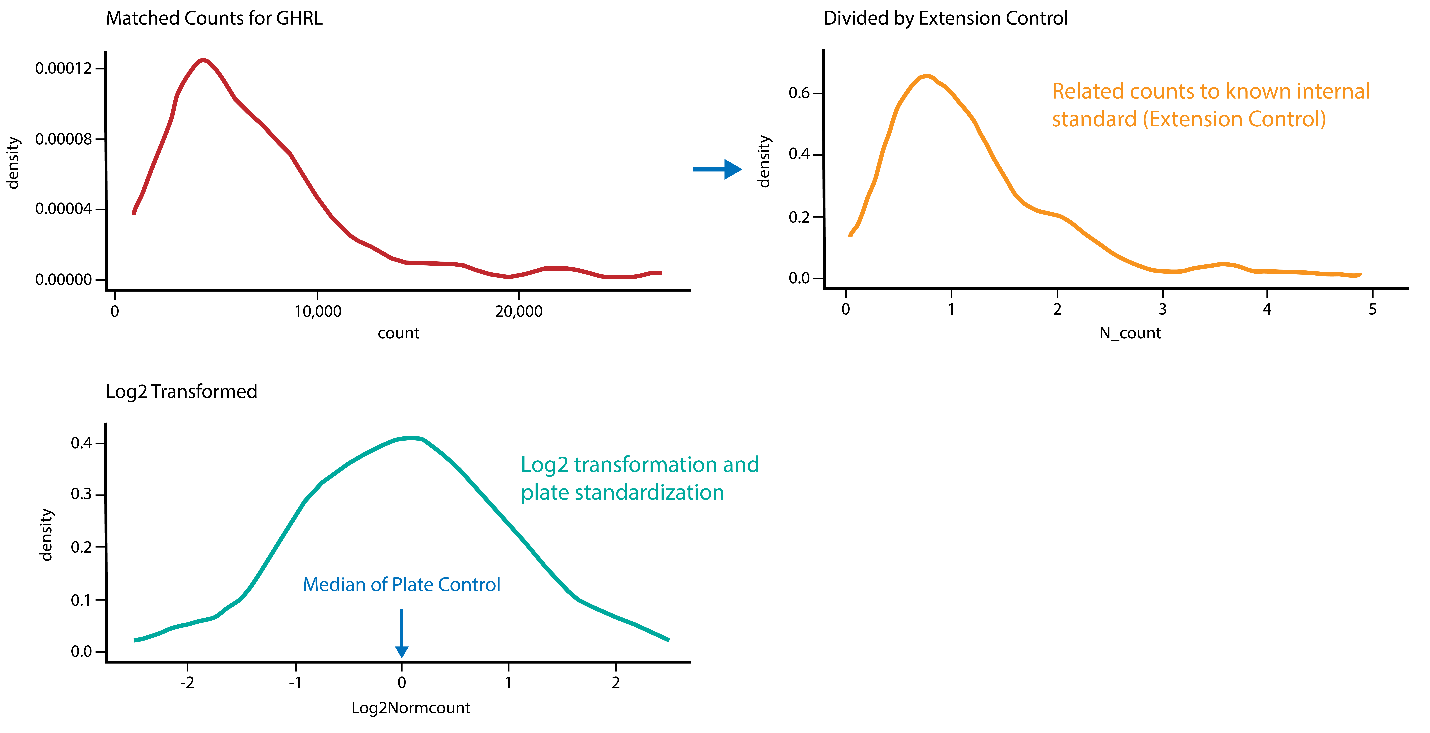
Figure 1: Summary of the three steps involved in NPX generation and data normalization. Step 1 is illustrated by the red graph, step 2 by the yellow graph, and step 3 (data normalization) by the turquoise graph. GHRL refers to appetite-regulating hormone (Uniprot ID: Q9UBU3).
AMP PD Quality Controls
LOD and CV Calculation
LOD is defined as being three standard deviations above the median NPX of negative controls. The median is set using all samples annotated as negative controls per plate. A predefined standard deviation is used (fixSD). Detectability is calculated per assay and plate and is defined by the percentage of samples above the LOD threshold. The overall detectability of the project is generated and reported in the CoA:
The CV is calculated per assay (i) using the assumption of a log-normal distribution. The average CV is then calculated across panels and included in the CoA output.
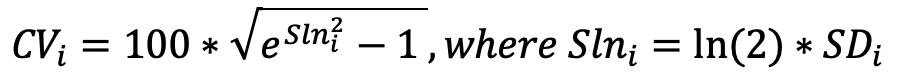
Bridging Datasets Methodology
Inclusion of Bridging Samples:
The final goal was to perform a joint analysis of datasets D01 and D02, and it was recommended to use common samples in the two batches that could help with batch correction to remove technical variation between the runs. Olink recommends the inclusion of 16-24 of these “bridging samples” across runs and the protein levels measured for these samples can be used as a common reference between batches, used to normalize and alleviate any potential bias. In this experiment, 39 samples from the D01 batch were included as bridging samples with the D02 dataset. These samples were selected using the dplyr R package to find the intersecting samples between D01 and D02 datasets. This list of overlapping samples was used as part of the olink_normalization_bridge_function from the Olink Analyze R package.
Bridging Data and QC
The bridging procedure involved the calculation of the median of the paired NPX differences per assay between the bridging samples. This is the assay specific adjustment factor which is used to adjust NPX values between all remaining samples in the two datasets. In this process, the D01 dataset was considered the reference dataset and its NPX values remained unaltered. The D02 dataset was adjusted to the reference dataset based on the adjustment factors.
In some cases, the bridging led to the duplication of the same sample x uniprot with two separate NPX values. In addition, there are cases in which there is a duplicate uniprot ID due to the same uniprot ID being in more than one Olink panel or there being two different Olink assays that map to that uniprot ID. In all of these cases, the mean NPX value was calculated and used in the final data product, resulting in one NPX value for every uniprot ID for every sample ID.
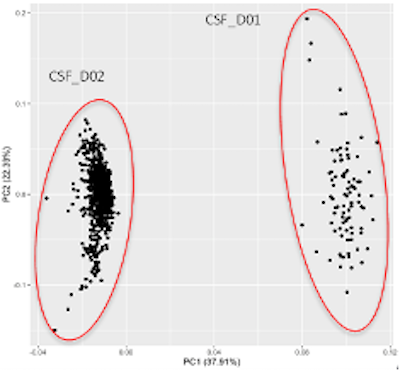
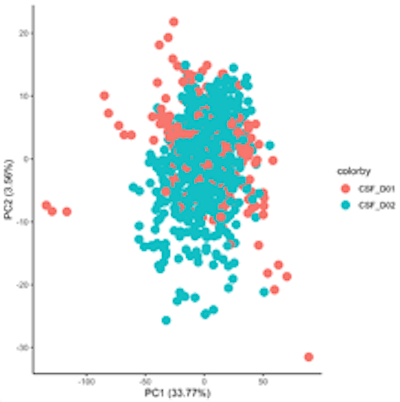
The bridging and mean calculations were all conducted using R and were performed separately for cerebrospinal fluid data sets and the plasma data sets. Results of the bridging can be seen below through PCA. Figure 1a shows dataset D01 and D02 prior to bridging. Separation along PC1 shows datasets D01 and D02 as distinct, with 37.91% of the variation due to batch effects. Figure 1b shows that after performing bridging, D01 and D02 no longer show separation along PC1, indicating that bridging was successful and batch effects were removed.